ABFE¶
This document describes how to run an ABFE simulation using Deep Origin tools.
The ABFE class runs an ordered sequence of workflow steps:
["system-prep", "abfe"]— protein + ligand through system prep, then FEP (default)["abfe"]— FEP only on an existingPreparedSystem
Prerequisites¶
We assume that you have prepared a protein, docked a ligand to it, and obtained a pose that you want to proceed with.
In this tutorial we will use a protein and ligand from the example dataset.
from deeporigin.drug_discovery import (
ABFE,
ABFEParams,
BRD_DATA_DIR,
Protein,
Ligand,
)
protein = Protein.from_file(BRD_DATA_DIR / "brd.pdb")
protein.sync()
ligand = Ligand.from_sdf(BRD_DATA_DIR / "brd-2.sdf")
ligand.sync()
For more details on how to get started, see Getting Started .
Combined workflow (recommended)¶
Pass protein and ligand to run system preparation and ABFE in one execution:
abfe = ABFE(protein=protein, ligand=ligand)
abfe.start(quote=True)
abfe.estimate
After quoting, confirm and watch progress as shown below. When the system-prep step completes, you can load the prepared system from the same execution:
prepared = abfe.get_prepared_system()
prepared.show()
Two-step alternative (SystemPrep)¶
You can still prepare the system separately with SystemPrep, then submit FEP-only
steps=["abfe"]:
from deeporigin.drug_discovery import SystemPrep, PreparedSystem
sysprep = SystemPrep(protein=protein, ligand=ligand)
system = sysprep.run()
system.show()
abfe = ABFE(prepared_system=system)
abfe.start(quote=True)
abfe.estimate
System prep builds two solvated models:
- Binding system — protein and ligand in explicit solvent (binding leg).
- Ligand solvation system — ligand alone in solvent (solvation leg).
You will see something like:
Estimating costs¶
Before starting an ABFE run, you can estimate costs using start(quote=True) on
either workflow entry point (combined or two-step):
abfe = ABFE(protein=protein, ligand=ligand)
abfe.start(quote=True)
abfe.estimate
You will get back an estimate (in USD) of how much this run will cost.
Starting an ABFE run¶
Confirming a quoted Job¶
To confirm the quoted price and start the execution, do:
abfe.confirm()
This will start the ABFE run. Use the following to monitor the status of the job:
task = await abfe.watch()
You will see a widget similar to this (progress simulated for demonstration):
Parameters¶
ABFE simulation parameters are controlled via the ABFEParams dataclass. Default values are tuned for production runs; most users will only need to adjust a small number of fields.
Viewing parameters¶
To inspect the current parameters on an ABFE object, access the params property:
abfe.params
Expected output
Parameters are printed one per line. Fields that have been changed from their defaults are marked with an asterisk (*):
ABFEParams(
annihilate: True
dt: 0.004
temperature: 298.15
cutoff: 0.9
repeats: 3 *
replex_period_ps: 2.5
test_run: 0
binding_n_windows: 48
binding_npt_reduce_restraints_ns: 2.0
binding_nvt_heating_ns: 1.0
binding_steps: 1250000
solvation_n_windows: 32
solvation_npt_reduce_restraints_ns: 0.2
solvation_nvt_heating_ns: 0.1
solvation_steps: 500000
)
Modifying parameters¶
ABFEParams is a frozen dataclass. Use dataclasses.replace() to build a modified copy and pass it to the constructor:
from deeporigin.drug_discovery import ABFEParams
from dataclasses import replace
params = replace(ABFEParams(), repeats=3, temperature=300)
params
Parameters modified from the defaults are indicated with an asterisk:
ABFEParams(
annihilate: True
dt: 0.004
temperature: 300 *
cutoff: 0.9
repeats: 3 *
replex_period_ps: 2.5
binding_n_windows: 48
binding_npt_reduce_restraints_ns: 2.0
binding_nvt_heating_ns: 1.0
binding_steps: 1250000
solvation_n_windows: 32
solvation_npt_reduce_restraints_ns: 0.2
solvation_nvt_heating_ns: 0.1
solvation_steps: 500000
)
Changing parameters may lead to simulation failures
Some parameters, such as dt, are constrained to specific ranges. You will not be allowed to start a simulation run if these parameters fall outside valid ranges.
Changing parameters away from their defaults may lead to simulation failures.
Results¶
Viewing results¶
After a run completes successfully, fetch results with:
df = abfe.get_results()
df
This shows a table similar to:
| dG | Std | AnalyticalCorr | Repeats | SMILES | r_exp_dg |
|---|---|---|---|---|---|
| -9.98 | 0.0 | -11.46 | 1 | COCCn1cc(-c2cccc(C(=O)N(C)C)c2)c2cc[nH]c2c1=O | -7.22 |
Viewing trajectories¶
To view MD trajectories from this run, refer to ABFE.
Viewing overlap matrix¶
An FEP overlap matrix is a diagnostic used in free energy perturbation calculations to evaluate how well neighboring λ states sample overlapping regions of configuration space. Each matrix element measures the statistical overlap between configurations from different states based on their energy distributions. The goal is to ensure every state has overlap with its neighbors in both directions – so that off-diagonal elements are sufficiently larger than zero.
The plot path is read from the data-platform result for this execution (default
repeat=1, same indexing as abfe.show_trajectory). In Jupyter:
abfe.show_overlap_matrix(run="binding")
An image such as the following will be shown:
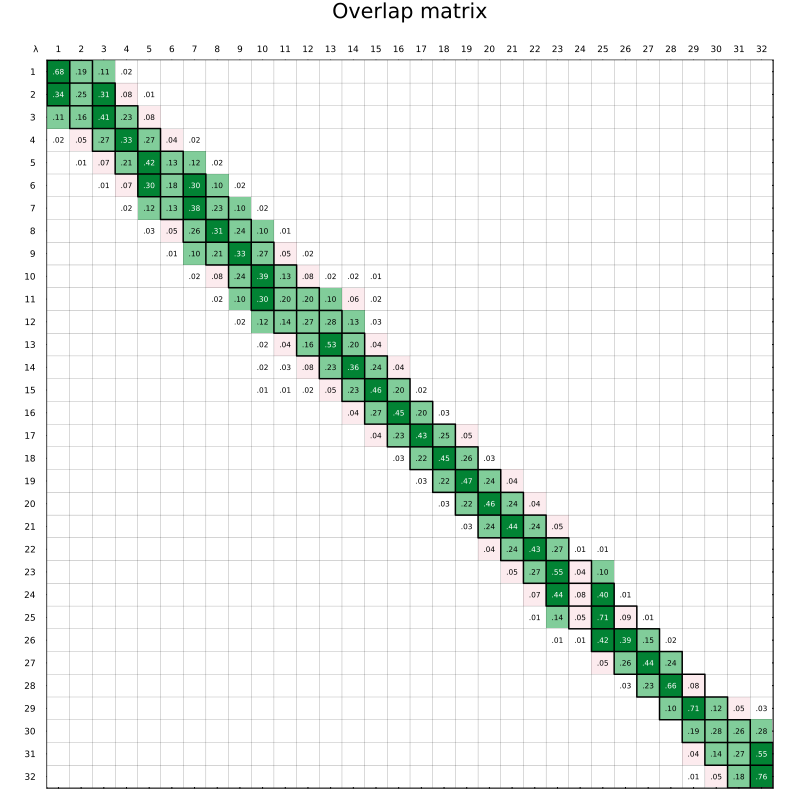
To view the overlap matrix for the solvation leg:
abfe.show_overlap_matrix(run="solvation")
Viewing convergence time plots¶
The time-convergence diagnostic (convergence_plot in the result payload) can
be shown the same way as the overlap matrix (default repeat=1, same as
abfe.show_overlap_matrix and abfe.show_trajectory):
abfe.show_convergence_time(run="binding")
An image such as the following will be shown:
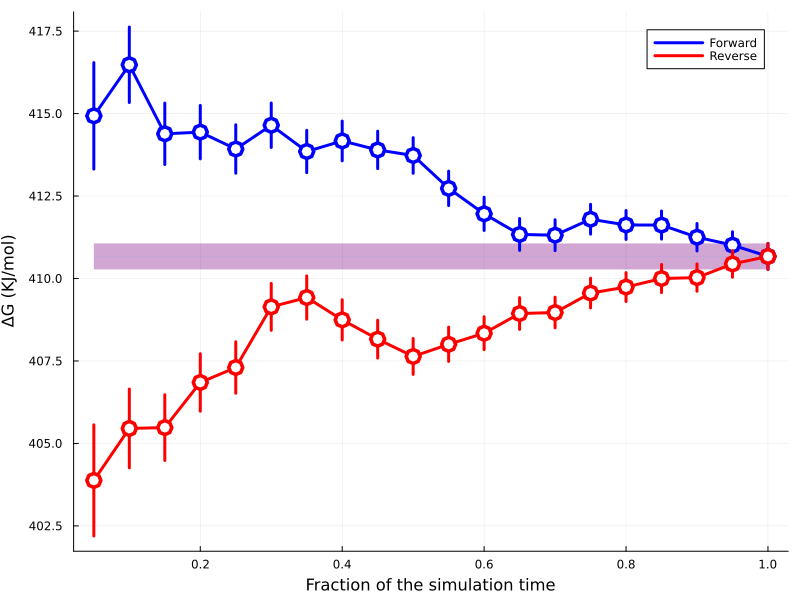
For the solvation leg:
abfe.show_convergence_time(run="solvation")